From What You Know From Lecture Another Organic Product Is Almost Certainly Formed
Rearrangements Induced by Cationic or Electron Deficient Sites |
|---|
Cationic Rearrangements
In the first half of the nineteenth century it was mostly believed that reactions of organic compounds proceeded with minimal structural change. This tenet simplified the elucidation of the numerous substitution, improver and elimination reactions that characterized the beliefs of mutual functional groups. Even so, subsequent discoveries showed that nature was not always so obliging, leaving chemists and chemistry students to grapple with the possibility of deep seated structural alter occurring during sure reactions. A large number of these structural rearrangements are triggered by intermediates incorporating positively charged or electron scarce atoms, which in the case of carbon are carbocations. Ii such examples, already noted, are the addition of HCl to 3,3-dimethyl-1-butene and forced hydrolysis of neopentyl bromide. This chapter will draw and talk over other cases of this intriguing group of transformations.
1. Wagner-Meerwein Rearrangements
The chemical behavior of neopentyl bromide, two,ii-dimethyl-ane-bromopropane, is an instructive place to begin this discussion. The very low SouthwardDue northii reactivity of this 1º-bromide was noted earlier, and explained past steric hindrance to the required 180º alignment of reacting orbitals. Under conditions that favor SNone reactivity, such as solution in wet formic acid, neopentyl bromide reacts at roughly the same rate every bit ethyl bromide. Both of these compounds are 1º-alkyl halides, and for an SouthwardN1 reaction the rate determining footstep requires ionization to a 1º-carbocation. As noted in the carbocation stability social club shown below, such carbocations are relatively unstable and are formed slowly. The production from ethyl bromide is ethanol, the simple and direct commutation product, just neopentyl bromide yields 2-methyl-2-butanol instead of the expected neopentyl booze. A change in the way the five carbon atoms in this product are bonded to each other has conspicuously taken place.
| Carbocation Stability | CH3 (+) | < | CH3CH2 (+) | < | (CH3)2CH(+) | ≈ | CHtwo=CH-CH2 (+) | < | C6H5CH2 (+) | ≈ | (CH3)3C(+) |
Once formed, the ethyl cation tin only exist transformed by a commutation or elimination process. In the case of the neopentyl cation, yet, the initially formed 1º-carbocation may be converted to a more stable 3º-carbocation past the ane,2-shift of an side by side methyl grouping with its bonding electrons. A mechanism demonstrating such a rearrangement is shown below, and it explains the overall structural changes very nicely.
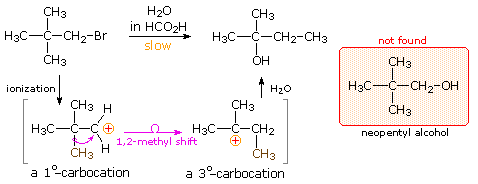
Increasing the stability of carbocation intermediates is not the only gene that leads to molecular rearrangement. If angle strain , torsional strain or steric crowding in the reactant structure may is relieved by an alkyl or aryl shift to a carbocation site, such a rearrangement is commonly observed. The following examples illustrate rearrangements induced past the strain in a small ring. Although a 3º-carbocation is initially formed, the bending and torsional strain of the four-membered band is reduced by a methylene group shift resulting in ring expansion to a 2º-carbocation. Clicking on the equation diagram will display a machinery for these transformations.
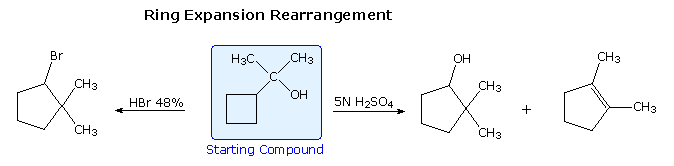
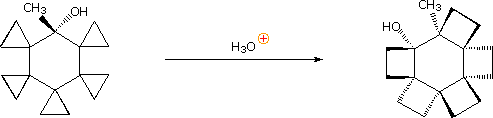
Post-obit the ring expansion step other reactions may take identify, depending on the conditions. In aqueous acid the rearranged 2º-carbocation may bond to a water nucleophile, producing a 2º-alcohol, lose a proton to water, giving 3,3-dimethylcyclopentene (not shown), or undergo a 2nd rearrangement to a 3º-carbocation, which and then forms i,2-dimethylcyclopentene. Indeed, it is not uncommon to encounter sequences of rearrangements in more complex compounds, and these may produce products with structures remarkably different from that of the starting compound. The post-obit equation shows i such reaction. A curved arrow representation of the five sequential ring expansion steps volition be added to the equation by clicking on the diagram.
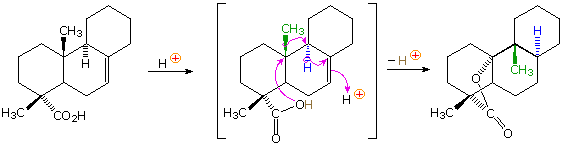
In the terminology of pericyclic reactions, one,2-alkyl shifts of this kind are classified as [1,2]-sigmatropic shifts. Since this is a 2-electron process (the 2 electrons in the relocated sigma bond), the rearrangement is predicted to be suprafacial. Considerable evidence supporting this conclusion has been obtained, every bit the post-obit example shows. Protonation of the double bond gives a 3º-carbocation. An adjacent hydrogen atom (colored blue) shifts as a hydride moiety to create a new 3º-carbocation, which in plough induces the shift of a methyl group (colored green) with germination of yet another 3º-carbocation. This electrophilic center and then bonds to the nucleophilic oxygen of the carboxylic acid function, releasing a catalytic proton to proceed the procedure. Because of the fused polycyclic structure of this chemical compound, the relative orientation of the migrating groups is easily adamant, and is seen to be suprafacial. Rearrangements consisting of consecutive 1,ii-shifts frequently take place in a concerted, and therefore stereospecific fashion; however, it must not be assumed that the group shifts are simultaneous. Each shift involves a separate transition land in which the positive charge is delocalized over the migration terminus, origin and migrating group.
Many of the nearly interesting rearrangements of this kind were discovered during structural studies of naturally occurring compounds. Among these the terpenes presented numerous remarkable reactions, and the names of two chemists who were instrumental in unraveling their circuitous transformations, H. Meerwein and Thou. Wagner, are permanently associated with these rearrangements. The add-on of gaseous HCl to α-pinene proved particularly puzzling to these early chemists. Under ordinary conditions, this liquid component of turpentine gave a crystalline C10H17Cl compound, originally called "artificial camphor", now known as bornyl chloride. An unstable isomer, pinene hydrochloride, can exist isolated under mild atmospheric condition, but it rapidly isomerizes to bornyl chloride. Treatment of bornyl chloride with base of operations gave a crystalline isomer of pinene called camphene, together with small amounts of another unsaturated hydrocarbon (bornylene). Addition of HCl to camphene, in a similar manner, initially produces an unstable chloride (camphene hydrochloride) which chop-chop isomerizes to isobornyl chloride, a stereoisomer of bornyl chloride. We at present know that bornyl chloride and isobornyl chloride are endo / exo-2-chloro isomers of the 1,7,seven-trimethylbicyclo[ii.two.1]bicycloheptane system. Structural formulas for these compounds are fatigued below, along with camphene, the rearranged emptying production.
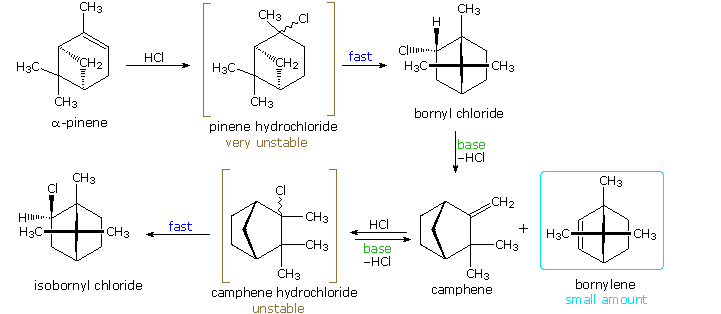
Mechanisms for these rearrangements will be pictured by clicking on the in a higher place diagram. In the new brandish we run into that both pinene and camphene form 3º-carbocations when the double bond is protonated. Rearrangement to a 2º-carbocation is favored by relief of modest-ring strain in the instance of pinene, and relief of steric congestion in the example of camphene. However, this is an oversimplification which ignores the fact that these reactions take place in nonpolar solvents, and are unlikely to involve detached, unassociated carbocations. Some of the stereoelectronic effects that influence these reactions will exist shown by clicking on the above diagram a 2nd time. Structures for the initially formed unstable hydrochlorides of pinene and camphene are drawn on the left. Optimal orbital overlap of breaking and forming bonds requires rear-side approach of the shifting alkyl group to the site of the leaving chloride anion, in a fashion like to a SouthwardN2 reaction. The chloride anion is located on i side of the carbocation formed past the alkyl shift, and immediately bonds to that face of the tricoordinate carbon. In this view of these rearrangements, the chloride anion never escapes the attractive influence of its cationic partner, and the production stereoselectivity is understandable. Lewis acid catalysts (east.k. FeCliii) catalyze these rearrangements, and the product favored at equilibrium is bornyl chloride.
The rearrangement that occurs nether base catalyzed elimination conditions reflects the eclipsed configuration of the two-carbon bridge bearing the chlorine cantlet. Considering of this configuration, the anti-coplanar construction favored by the E2 transition land cannot be accomplished. Syn elimination gives a pocket-size corporeality of bornylene, just rearrangement to a camphene forerunner predominates. Repeated clicking on the to a higher place diagram will cycle the displays.
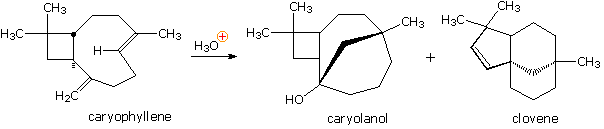
Withal some other example of the remarkable acid-catalyzed rearrangements found to occur with terpenes was observed in a study of the sesquiterpene caryophyllene (from oil of cloves). Hither it is evident that reactive sites may interact and class bonds from one side of a medium-sized ring to another side. The mechanisms for many such rearrangements accept been, and even so are studied with great involvement.
2. Pinacol Rearrangement
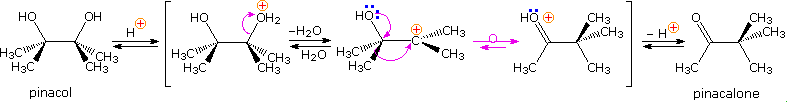
The pinacol rearrangement was the first molecular rearrangement identified as such by early chemists. The defining example of a pinacol rearrangement is shown in the post-obit diagram. Pinacol itself is produced by magnesium reduction of acetone, probably past mode of a ketyl intermediate. Since the diol is symmetrical, protonation and loss of water takes place with equal probability at either hydroxyl group. The resulting 3º-carbocation is relatively stable, and has been shown to return to pinacol by reaction in the presence of isotopically labeled water. A 1,two-methyl shift generates an even more stable carbocation in which the charge is delocalized by heteroatom resonance. Indeed, this new cation is just the conjugate acrid of the ketone pinacolone, which is the product of repeated rearrangements catalyzed by proton transfer. Each step in this rearrangement is potentially reversible, as demonstrated by the acrid catalyzed dehydration of pinacolone (and pinacol) to ii,iii-dimethyl-1,3-butadiene nether vigorous conditions.
Many factors must be considered when analyzing the form of a pinacol rearrangement. These include:
• Which hydroxyl group is lost every bit water? or Which intermediate carbocation is more stable?
• What is the inherent shifting tendency (migratory bent) of different substituent groups?
• What is the influence of steric hindrance and other strain factors on the rearrangement?
• Are epoxides formed as intermediates in the pinacol rearrangement?
• Does production stability govern the issue of competing rearrangements?
• Practise the reaction weather (i.e. type of acrid, concentration, solvent and temperature) influence the course of rearrangement?
Virtually all of these factors take been shown to be of import in one or more cases, and a total assay of their circuitous interaction is beyond the scope of this text. Nevertheless, a few examples will be presented to demonstrate the full general nature of this transformation, and to illustrate the action of some of the above factors. In the kickoff reaction shown below, we come across an example of kinetic versus thermodynamic product control. Under mild acid handling, the diol rearranges rapidly to an aldehyde by way of a i,2-hydrogen shift to the initially formed diphenyl 3º-carbocation. More than vigorous acid treatment of the diol or the aldehyde generates the more stable phenyl ketone (conjugation of the phenyl and carbonyl groups). Mechanisms for this and the other reactions will be presented by clicking on the diagram. A pink colored arrow designates rearrangement; calorie-free blue arrows indicate epoxide band closing or opening reactions. Repeated clicking toggles the reaction and mechanism displays.
The second example describes a similar reacting system, which provides additional information from stereochemical and isotopic labeling features. Loss of h2o from the 3º-carbinol site, followed past a reversible 1,2-hydride shift, generates the conjugate acid of the ketone product. At short reaction times, racemization of recovered diol starting material occurs at the aforementioned rate as rearrangement. A corresponding phenyl shift to the initially formed 3º-carbocation generates the aldehyde conjugate acrid, and the aldehyde itself has been shown to isomerize to the aforementioned rearranged ketone under the weather condition of this pinacol rearrangement. An isotopic carbon label (colored green) in either the diol or aldehyde is scrambled (colored brown) in the course of these reactions, suggesting an epoxide intermediate.
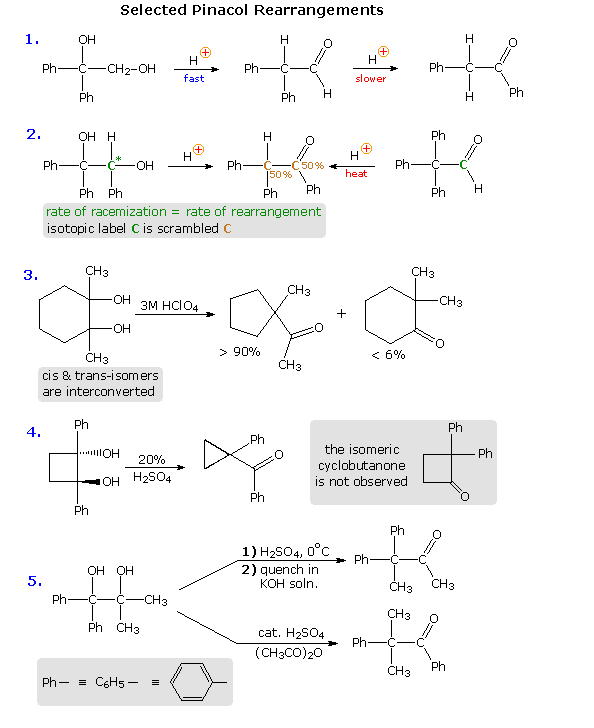
In reaction # 3 either the cis or trans diol may be used as a reactant. These isomers are rapidly interconverted under the rearrangement conditions, indicating that the initial water loss is reversible; a upshot confirmed by isotopic oxygen exchange. The clear preference for a methylene group shift versus a methyl group shift may reverberate inherent migratory aptitudes, or possibly group configurations in the 3º-carbocation intermediate. In the conformation shown here both methyl and methylene groups may shift, or an epoxide band may be formed reversibly. An alternative chair-like conformation having an equatorial methyl grouping should be more stable, just would not be suitable for a methyl shift. The predominant ring contraction is therefore understandable. Reaction # 4 is an unusual case in which a strained ring contracts to an even smaller ring. Phenyl groups by and large accept a high migratory bent, then the failure to obtain 2,ii-diphenylcyclobutanone as a production might seem surprising. However, the carbocation resulting from a phenyl shift would exist merely as strained every bit its precursor; whereas the shift of a ring methylene grouping generates an unstrained cation stabilized past phenyl and oxygen substituents. Conjugative stabilization of the phenyl ketone and absence of sptwo hybridized carbon atoms in the small band may also contribute to the stability of the observed product.
Finally, reaction # 5 clearly shows the influence of reaction weather condition on product composition, simply explaining the way in which different conditions adjy the outcome is challenging. Treatment with cold sulfuric acrid should produce the more stable diphenyl 3º-carbocation, and a methyl group shift would and then lead to the observed production. The action of a Lewis acid in acetic anhydride, on the other hand, may selectively acetylate the less hindered dimethyl carbinol. In this event the acetate becomes the favored leaving group (presumably coordinated with acid), followed by a 1,2-phenyl shift. It is reported that the symmetrically substituted isomeric diol (drawn in the shaded box) rearranges exclusively by a methyl shift, only the configuration of the starting material was not stated (ii diastereomers are possible).
Because of the influence of other factors (to a higher place), it has not been possible to determine an unambiguous migratory club for substituents in the pinacol rearrangement. Withal, some full general trends are discernible. Benzopinacol, (C6H5)2C(OH)C(OH)(C6H5)2, undergoes rapid rearrangement to (C6Hv)threeCCOC6H5 under much milder weather than required for pinacol. Indeed, it is often the example that phenyl or other aromatic substituents side by side to a forming carbocation will facilitate that ionization in the form of their migration to the cationic site. In non-aromatic compounds of the blazon (CH3)2C(OH)C(OH)RCH3, migration of R increases in the manner R = CH3 < R = C2H5 << R = (CH3)3C. Since the shifting alkyl group must comport function of the overall positive accuse, alkyl substitution should accept a stabilizing influence on the rearrangement transition country. Finally, fluorine substitution, every bit in Chalf dozenH5(CFthree)C(OH)C(OH)(CF3)CsixH5 renders the diol unreactive nether acid catalyzed rearrangement weather condition. Here, the powerful inductive withdrawal of electrons past fluorine inhibits positive accuse formation.
three. Tiffeneau-Demjanov Rearrangement
Ambiguity in determining the initial site of carbocation formation presented a problem in the assay of many pinacol rearrangements. This uncertainty can be removed past nitrous acrid deamination of the respective 1º-aminoalcohols, equally shown in the following equation. Since this reaction is normally carried out under very mild conditions, the possibility that subsequent transformations may obscure the initial rearrangement is reduced considerably.
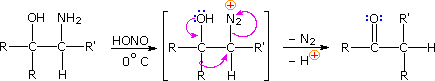
The Tiffeneau-Demjanov rearrangement is often used to transform a cyclic ketone into a homologue that is one band size larger. Such an application, which proceeds past mode of a cyanohydrin intermediate, is shown in the first example below. Cyclic ketones have two blastoff-carbon atoms, each of which might shift to the nascent 1º-carbocation. If R = H in the case shown here, these two groups are identical and on shifting give the same product. If R = CH3, the 2º-alkyl grouping shifts preferentially, the chief product being 3-methylcyclohexanone; the 2-methyl isomer is a minor product. The second reaction is informative because it demonstrates that the chiral 2º-butyl grouping moves with retention of configuration.
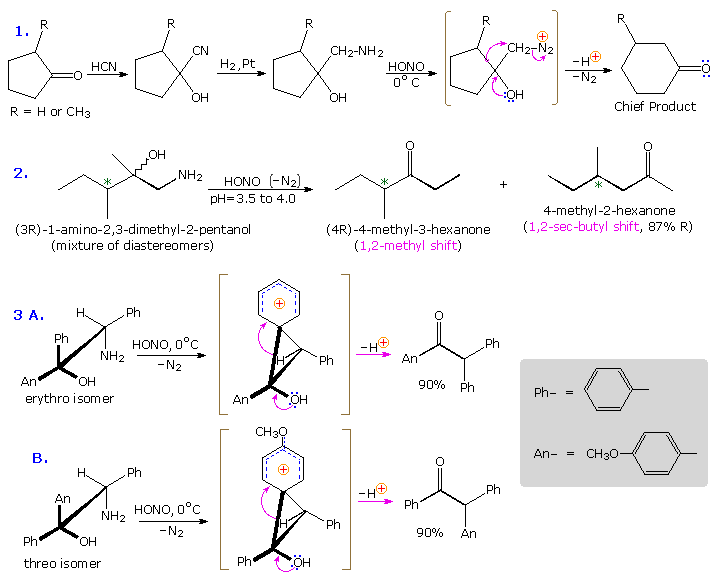
The third example illustrates the importance of substrate configuration on the course of rearrangement. The initial stage of an aryl group shift to an side by side carbocation site may be viewed equally an intramolecular electrophilic commutation of the Friedel-Crafts type. Aryl band approach from the side contrary to the departing nitrogen of the diazonium ion generates a phenonium ion intermediate (shown in brackets higher up), the construction of which is similar to a benzenonium ion. In these ii examples, diastereomeric reactants lead preferentially to diastereomeric intermediates, fifty-fifty though the anisyl grouping has a much greater migratory bent than phenyl. Electron pair donation by the hydroxyl substituent then acts to open up the 3-membered ring of these intermediates, yielding the ketone products.
4. Anchimeric Assistance
When the solvolysis rates of alkyl halides and sulfonate esters are measured, some curious influences of neighboring substituents are observed. For case, ethyl chloride, neopentyl chloride (2,two-dimethylpropyl chloride) and two,2,2-triphenylethyl chloride are all 1º-alkyl chlorides, which hydrolyze in wet formic acid to mixtures of alcohols and olefins (SouthwardN1 & E1 mechanisms). The reaction rates for ethyl chloride and neopentyl chloride are nearly identical, just the triphenyl compound reacts threescore,000 times faster. Equations for the latter 2 solvolyses are shown in the following diagram. It is apparent that in both cases an initially formed 1º-carbocation has rearranged prior to product formation, equally depicted by clicking on the diagram. However, the increased rate of the phenyl substituted compound is perplexing, specially in view of the greater electronegativity of phenyl groups relative to methyl (annotation that diphenylacetic acid is over nine times more acidic than isobutyric acid). To explain the unexpected reactivity of two,two,2-triphenylethyl chloride it is proposed that the pi-electrons of a suitably oriented phenyl group assist the 1º-chloride ionization by bail formation from the side opposite the C-Cl bail, every bit shown past clicking on the diagram a second time. This intramolecular interaction corresponds to the last example in the previous section, and is similar to an intramolecular SouthNii reaction. The resulting phenonium ion would immediately open to a 3º-carbocation, in which the assisting phenyl group has shifted to an adjacent position. In this style a neighboring aromatic ring accelerates the rate-determining (endothermic) ionization pace, an influence called anchimeric assistance (Greek: anchi = neighbour).
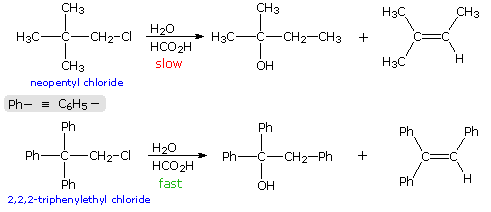
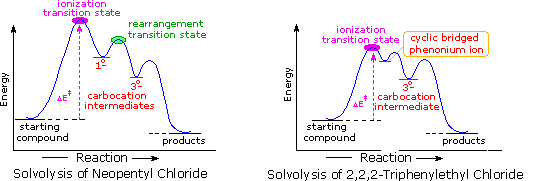
The post-obit energy profiles for these reactions illustrate the sequence of events. Both reactions begin by an initial rate-determining ionization step, the transition country of which is colored pink. The activation energy for this pace is larger for neopentyl chloride because it leads to a detached 1º-carbocation. On the other hand, the ionization of triphenylethyl chloride proceeds with assistance from a neighboring phenyl group, and the resulting phenonium ion immediately opens to a very stable diphenyl 3º-carbocation. The second step in the neopentyl chloride solvolysis is a rapid rearrangement of the 1º-carbocation to an isomeric 3º-carbocation. The transition state for this rearrangement is colored green. In both cases, the 3º-carbocation intermediate finally disproportionates to a mixture of commutation and elimination products. The essential divergence is that the ionization transition country for neopentyl chloride suffers all the disadvantages associated with the generation of a 1º-carbocation; whereas, the transition state for ionization of triphenylethyl chloride is lowered in energy by its phenonium-like character.
Anchimeric help not only manifests itself in enhancement of ionization, just also influences the stereochemical outcome of reactions. The acetolysis of diastereomeric 3-phenyl-2-butanol derivatives provides an example. This alcohol has two chiral centers, and therefore has iv stereoisomers in the form of two pairs of enantiomers. The diastereomeric configurations are chosen erythro and threo, co-ordinate to their correlation with the tetroses erythrose and threose. As a rule, erythro isomers may assume an eclipsed conformation in which identical or like substituents on the two stereogenic sites eclipse each other. Threo isomers cannot assume such a conformation. In the following diagram, a tosylate derivative of one enantiomer of each diastereomer is drawn as a Fischer projection. These isomers were solvolyzed in hot acetic acid solution, buffered with sodium acetate, and the configurations of the resulting acetate esters were determined. As expected from a SNorthward1 process, some E1 emptying production was besides obtained. Remarkably, each diastereomer is converted to its equivalent diastereomeric acetate (retention of configuration). Furthermore,the erythro compound retains its enantiomeric purity; whereas the threo tosylate gives racemic acetate and is itself racemized during reaction. If an open carbocation intermediate were formed in these reactions, mixtures of erythro and threo acetates would be expected from both tosylates, but just trace amounts of the opposite diastereomer were institute among the products.
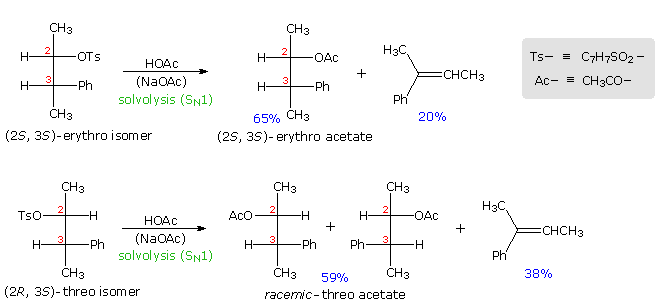
By clicking on the diagram the controlling influence of phenyl group anchimeric assistance volition exist demonstrated. First, the molecule assumes a conformation in which the phenyl substituent is oriented anti to the tosylate group. Next, a pair of pi-electrons from the benzene ring bonds to C2 every bit the tosylate anion departs, generating a phenonium intermediate (in brackets). The intermediate from the erythro tosylate is chiral, but that from the threo tosylate is achiral (note the plane of symmetry bisecting the three-membered ring). In each case C2 & C3 are constitutionally equivalent, and nucleophilic attack by acetate anion takes identify equally well at either position (green and calorie-free blue arrows). As a upshot of equal rates of production formation by acetate bonding to C2 & C3, the achiral threo intermediate yields a l:50 (racemic) mixture of threo enantiomers: (2R,3S) from the blueish arrows and (2S, 3R) from the green arrows. In dissimilarity, acetate bonding to C2 & C3 of the erythro intermediate produces the same enantiomer of the erythro product (2S,3S). Since the initial ionization to phenonium intermediates is reversible, nosotros are not surprised to find that unreacted erythro tosylate is unchanged; whereas, unreacted threo tosylate is racemized.
5. Anchimeric Assist by Other Neighboring Groups
The ability of the pi-electrons in a suitably oriented, neighboring benzene ring to facilitate C-X ionization, where X is a element of group vii or a sulfonate ester, was described in the previous department. Other aromatic rings, such every bit naphthalene, furan and thiophene, may function in a similar way, as may the pi-electrons of double and triple bonds. The following diagram shows three examples of neighboring double bond interaction, the first existence 1 of the most striking cases of anchimeric assistance on record. The use of dashed lines to show charge delocalization is a mutual do. The text box below the diagram provides additional commentary concerning these examples.
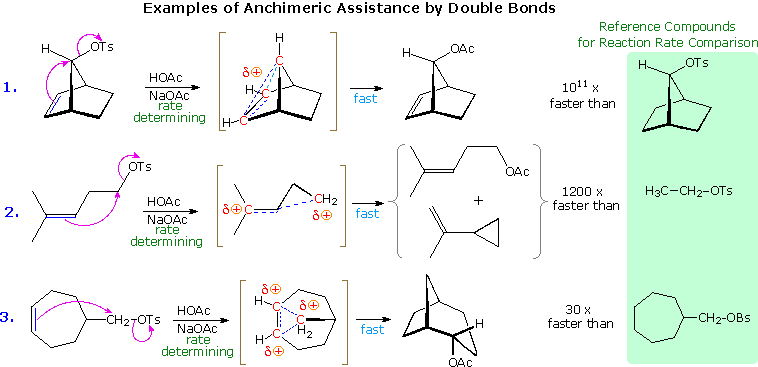 | |||||
| Neighboring | Double Bonds | Triple Bonds | Sulfur Atoms | Oxygen Atoms | Nitrogen Atoms |
|---|---|---|---|---|---|
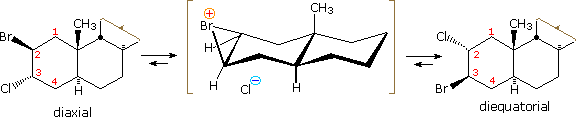
Examples of other neighboring group perturbations, including non-bonding electron pair assistance by neighboring sulfur, oxygen and nitrogen atoms volition be displayed to a higher place past clicking the appropriate button under the diagram. The text box commentary will change to suit the examples. In almost of the cases involving heteroatom assistance, an "onium" intermediate is formed, in which the heteroatom is charged. Adjacent halogen atoms may likewise stabilize carbocations, every bit noted before with respect to trans-anti additions to circadian alkenes. Functional rearrangement by way of halonium intermediates has besides been reported. For example, a chloroform solution of the diaxial 2-bromo-three-chlorosteroid, shown on the left below, spontaneously rearranges to the more stable diequatorial ii-chloro-three-bromo isomer drawn on the right. The rearrangement is reversible and proceeds by way of the circadian bromonium ion written in brackets.
6. The Nonclassical Carbocation Hypothesis
The function of carbocation intermediates in many organic reactions is well established. Some, such as tert-butyl, are localized. Some,such every bit allyl and benzyl, are stabilized by conjugation to pi-electron systems. Some, equally described above, are stabilized past bridging to neighboring nucleophiles. In all cases of anchimeric aid described in a higher place, a accuse delocalized or redistributed species is an intermediate on the reaction path. Such intermediates can be isolated in some cases, but they usually have only transitory being. The rate dispatch of ionization is attributed to structural and energetic similarities of the transition states to the intermediates they produce (the Hammond postulate).
Anchimeric aid is ordinarily associated with one or more of the following observable characteristics.
• Rate acceleration compared with similar reactions defective assistance.
• Stereoelectronic command that results in rate and product differences between stereoisomers.
• Retention of configuration in substitution products.
• Racemization of products (and oft reactants) when a symmetrical bridged intermediate is involved.
Solvolysis of the exo and endo-two-norbornyl sulfonate esters disclosed differences that suggested anchimeric assist for the exo-isomer. As shown in the following diagram, the rate of acetolysis of the exo-isomer is essentially faster than that of the endo-isomer, which reacts at a rate similar to the cyclohexyl derivative. The old exchange proceeds with complete retentiveness of configuration and racemization; whereas the endo-isomer is substituted with inversion of configuration and retains a small degree of optical activity. The source of this aid was proposed to be the electron pair of the C1 : C6 sigma bail, which is ideally oriented anti to the sulfonate leaving group. A sigma-delocalized ion (drawn in brackets), was proposed every bit an intermediate, displayed by clicking on the diagram. Since this bridged ion is symmetrical, formation of racemic acetate is expected. The term "nonclassical" was practical to this charge delocalized cation, inasmuch as it appeared to be unique.
By comparison, the endo-isomer ionizes to a classical 2º-carbocation, which is rapidly converted to the more stable nonclassical ion. Some acetate anion may bail to the 2º-carbocation before it changes, bookkeeping for the rest optical action in this reaction.
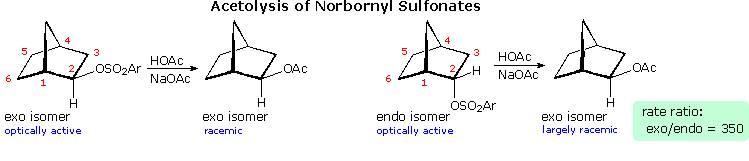
Non everyone was convinced by this interpretation of the bear witness. The primary protagonists favoring the nonclassical view were Southward. Winstein and J. D. Roberts. The primary opposition came from H. C. Chocolate-brown, who espoused a more conventional rationalization. Brown pointed out that the norbornyl compounds are meliorate compared with cyclopentyl than with cyclohexyl analogs (eclipsing strain), and in such a comparing the endo isomer is abnormally slow, the exo isomer being only 14 times faster than cyclopentyl. The racemic production was explained by bold the interconversion of enantiomeric classical carbocations was very rapid on the reaction fourth dimension scale. Brown also noted that attachment of a stabilizing aryl substituent at C2 did not reduce the rate enhancement of exo-ionization or the preference for exo-production formation. Since these latter solvolyses proceed by way of a benzylic cation, sigma-bail aid was assumed to be minimal. Consequently, charge per unit enhancement and retention of configuration become less pregnant as nonclassical indicators. This latter experiment, in which the aryl substituent was p-anisyl (An), is depicted on the left side of the diagram beneath.
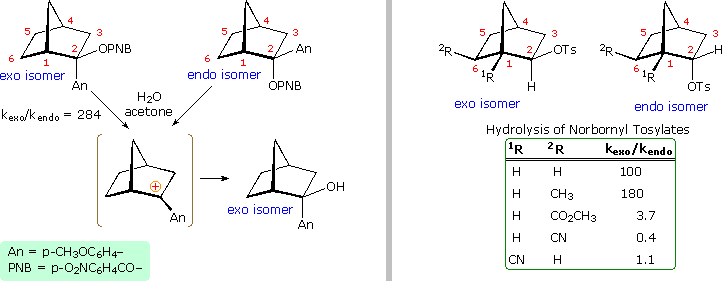
Despite Brown's damaging arguments, other experiments provided boosted support for the nonclassical view. As shown on the right side of the diagram, electron withdrawing substituents on C6 (2R) retarded exo-reactivity more than severely than endo-reactivity. A similar consequence was noted for such substituents at C1 (1R). This influence is best explained by the nonclassical hypothesis, in which partial positive accuse must be carried by C1, C2 & C6.
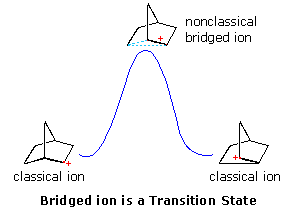
Interpretations of the considerable body of bear witness amassed at this betoken may be summarized in the diagram on the right. In the first brandish, the nonclassical bridged cation is shown as a transition land for the interconversion of the classical carbocations. A relationship of this kind corresponds to the rearrangement of neopentyl chloride. A 2d possibility, presented by clicking on the diagram, has the nonclassical ion as a college free energy intermediate, linking the classical ions. Finally, past clicking on the diagram a second time, the possibility that the nonclassical ion represents the more stable intermediate is drawn.
Past the mid 1960'south chemical and nmr techniques had improved to a phase that allowed direct observation of carbocations in low nucleophilic, acidic solutions, often referred to as "super acids". Much of this work was conducted by George Olah (Nobel Prize, 1994), using mixed solvents composed of SbF5, SO2, SOiiFtwo & And so2FCl. At depression temperatures, 1H and 13C nmr spectra of (CH3)3C(+) and (CH3)2CH(+) were obtained and interpreted. As anticipated, the charged tricoordinate carbon atom exhibited a 13C signal over 300ppm downfield from TMS. When similar nmr measurements were applied to the two-norbornyl cation, a number of fast proton shifts were disclosed. These could be "frozen out" by working at low temperature, the three,two-shift at -70º C and a faster 6,ii-shift at -130º C. The resulting spectrum, which remained unchanged at temperatures as low every bit -160º C, had no low field signals near that expected for a classical 2º-carbocation, and was supportive of the nonclassical construction. Recently, a solid state xiiiC nmr spectra at 5º Thousand proved consequent with the nonclassical ion. From these and other spectroscopic studies, the sigma-bridged nonclassical cation has been firmly identified as the more stable carbocation species having the ii-norbornyl structure. Farther confirmation was provided in 2013 past researchers in Germany, employing careful X-ray crystallographic measurements of an annealed [CviiHeleven]+[AltwoBr7]– salt at 40º K.
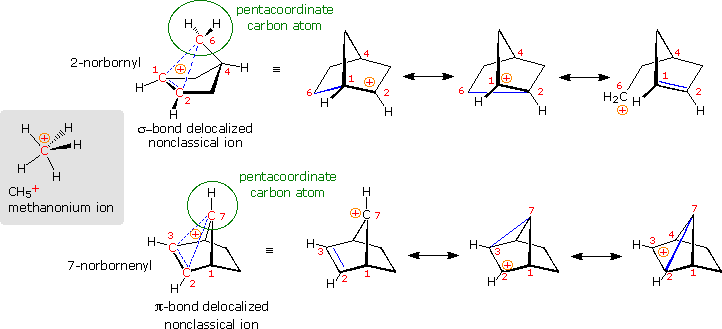
Are in that location other relatively stable nonclassical carbocations? Several that seem to fit this classification have been identified, but few accept been equally exhaustively studied every bit the two-norbornyl. 1 of the best criteria for evaluating candidate ions is to institute whether ane or more of the participating carbon atoms is hypervalent (has more 4 analogous groups). In the following diagram, the simplest hypervalent carbocation, methanonium, is drawn on the left in the gray shaded box. This ion is normally seen in the mass spectrum of methane (gas phase), but decomposes in solution as a consequence of its farthermost acidity. To its right are two larger non-classical ions, 2-norbornyl and 7-norbornenyl. A pentacoordinate carbon atom is identified in each case. Resonance contributors to these ions are shown to the right of the dashed bond representation, and in all the drawings the delocalized electron pair is colored blue. Finally, a broad overview of this classification, offered by Olah in his Nobel lecture, will be displayed by clicking on the diagram.
To run across a model of the two-norbornyl cation .
Rearrangements to Electron Scarce Heteroatoms |
|---|
Rearrangements to Electron Deficient Heteroatoms
1. Rearrangements of Cationic Oxygen
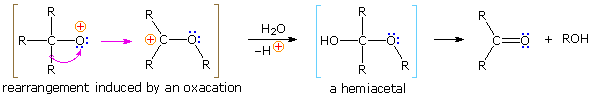
Protonation of the divalent oxygen atom of alcohols and ethers past strong acids produces a tricoordinate oxonium cation. Because the oxygen of an oxonium ion has a valence shell octet, it does not constitute an electron deficient site and cannot serve every bit a rearrangement terminus. To induce rearrangement in the same style as a tricoordinate carbocation, oxygen must be converted to a unicoordinate oxacation, as noted in the following diagram. A 1,2-alkyl or aryl shift then transforms a relatively unstable oxacation into a more stable carbocation.
The simplest precursor of an oxa cation is a peroxide or equivalent derivative (eastward.yard. R-O-OH or R-O-X). Removal of hydroxide anion from a hydroperoxide is energetically unfavorable, unless it is initially converted to a better leaving grouping in a manner similar to that used to facilitate substitution reactions of alcohols. By protonating the hydroxyl group, the leaving grouping becomes water, thus generating an oxacation. A useful industrial process for preparing phenol (and acetone) is based on this strategy.
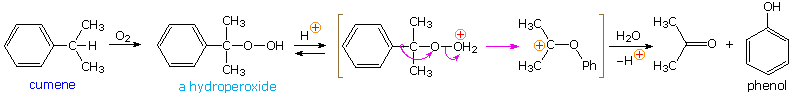
Baeyer-Villiger Rearrangement
The acid-catalyzed reaction of ketones with hydroperoxide derivatives is known every bit the Baeyer-Villiger reaction. A full general equation illustrating this oxidation reaction is shown beneath, and it may be noted that the rearrangement step is similar to that of a pinacol rearrangement. Esters or lactones are the chief products from ketone reactants. In this equation a detached oxacation is drawn as an intermediate, but it is more probable that the rearrangement is concerted, every bit will exist shown by clicking on the equation. In one case the peracid has added to the carbonyl group, the rearrangement may exist facilitated by an intramolecular hydrogen bond, in the style depicted in brackets on the right.
The migratory bent of various substituent groups (e.yard. 1R & 2R) is generally: 3º-alkyl > 2º-alkyl ~ benzyl ~ phenyl > 1º-alkyl > methyl. Stereoelectronic factors favor an anti-periplanar orientation of the migrating grouping to the leaving moiety, and volition command the rearrangement in some cases. An case volition exist displayed below on clicking the display a 2nd time. Peracid commutation with peracetic acid leads to an intramolecular Baeyer-Villiger reaction past mode of the bicyclic acylperoxide fatigued in brackets. Hither stereoelectronics favor migration of the less substituted α-carbon. The lactone production was identified by esterification and ester exchange with methanol to requite methyl 2-carbomethoxy-7-hydroxyheptanoate.
Aldehydes are unremarkably oxidized to carboxylic acids under the conditions used for the Baeyer-Villiger reaction.
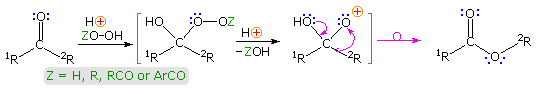
Although hydrogen peroxide itself may be used in the Bayer-Villiger reaction, it may add at both ends to reactive carbonyl groups, producing cyclic dimeric, trimeric and higher addition compounds. Consequently, derivatives such as peracids (Z = RCO & ArCO in a higher place) are the preferred reagents for this reaction. Amongst the most common peracids used in this respect are: peracetic acid, perbenzoic acid & meta-chloroperbenzoic acid (MCPBA). Four examples of this oxidative rearrangement are given in the following diagram. In almost of these examples the migrating grouping retains its configuration in the course of the rearrangement, as expected for a concerted process. In instance #3 it is interesting that migration of the bridgehead 3º-alkyl grouping is preferred over a possible phenyl shift.
Some Baeyer-Villiger Oxidative Rearrangements

2. Rearrangements of Cationic or Electron Scarce Nitrogen
Since many simple nitrogen compounds are bases, they form "onium" cations when protonated. 2 such cations are shown on the left (in the blue box) below. Because ammonium and iminium cations accept a nitrogen valence beat electron octet, such a nitrogen atom cannot serve as a site for nucleophile bonding or a migration terminus. For a nitrogen cation to initiate rearrangement it must have an unfilled valence vanquish, and two such azacations are shown in the center of the diagram (pink box). An electron deficient nitrogen atom does not have to exist a cation, as the nitrene example on the right (green box) demonstrates.
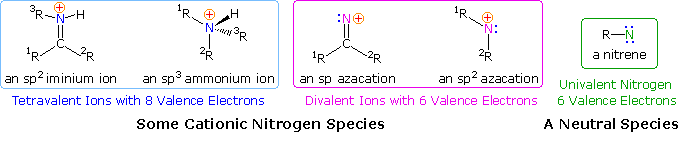
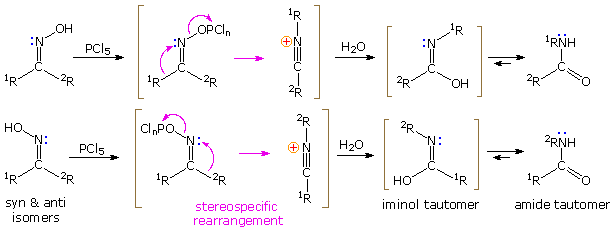
The Beckmann Rearrangement
If the hydroxyl group of a ketoxime is removed by the activeness of strong acrid or phosphorous pentachloride, followed by hydrolysis, an amide is formed. Consummate removal of the derivatized hydroxyl grouping and its bonding electron pair would generate a divalent sp-hybridized azacation of the type depicted in the previous diagram. Were this to occur, both carbon substituents (1R & twoR) would be candidates for the subsequent 1,2-shift. In practise, yet, information technology is always the grouping anti to the departing OH that migrates to nitrogen. This stereospecificity indicates that the 1,2-shift is concerted with N-O cleavage, as shown below. The resulting North-alkylated nitrilium intermediate volition react with nucleophiles (east.thousand. water) at the electrophilic carbon cantlet adjacent to the "onium" nitrogen. Annotation that the construction fatigued for this intermediate is the more favored of ii resonance contributors, inasmuch every bit all heavy atoms have filled valence shell octets. Bonding of a nucleophile to the nitrogen atom would require expanding its valence shell to include ten electrons, or formation of an unstable dipolar species. The initial production from hydration at carbon is an iminol, which immediately tautomerizes to the more stable amide. Reactions with PClv probably give an iminochloride, and this in turn is hydrolyzed to the same amide.
The first instance in the following grouping of reactions is a typical Beckmann rearrangement. The oxime from cyclohexanone has identical carbonyl substituents. and therefore exists as a single isomer. The product of the rearrangement is a lactam (a circadian amide), which can be hydrolyzed to an omega-amino acid. This lactam serves as an of import industrial precursor to nylon vi. The second example involves an oxime derivative with different carbonyl substituents, which exists every bit a pair of stereoisomers (syn & anti). The anti isomer rearranges past a 1,2-phenyl shift, whereas the syn isomer undergoes a 1,2-isopropyl shift. The former reaction is much faster than the latter, presumably because it proceeds by way of a relatively stable phenonium ion intermediate (structure in shaded box). Note that the picrate leaving group (2,four,half dozen-trinitrophenolate) is a stable anion. Example #three is another case that demonstrates the stereospecificity of the Beckmann rearrangement. The i,2-shift of the ortho-phenol substituent is faster than that of the unsubstituted phenyl grouping, and the hydroxyl is ideally located to bond to the electrophilic carbon of the intermediate. Consequently, the production from the anti isomer is a benzoxazole heterocycle.
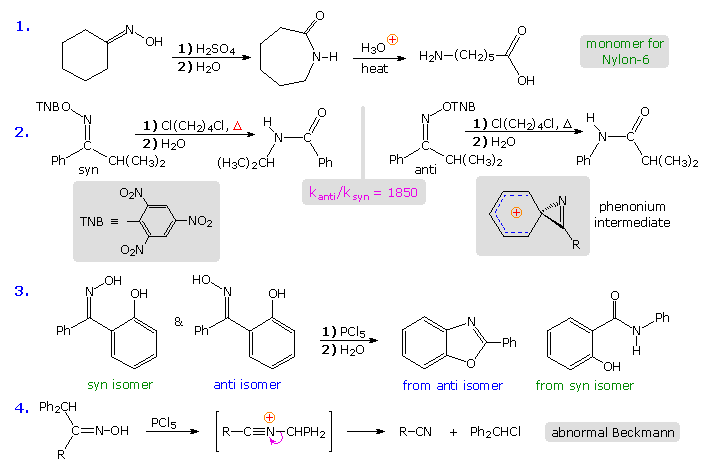
The quaternary example above shows an unusual difference in behavior that sometimes occurs when the migrating substituent fragments from the intermediate, leaving a nitrile as a stable product. This has been called an abnormal Beckmann reaction.
The rigid configuration of the phenonium cation shown above imposes a structural constraint that is nicely demonstrated by the rates of rearrangement of some fused ring bicyclic compounds. Clicking on the diagram will bear witness the results of such a study. Oxime derivatives of phenyl ketones incorporated in six, vii and eight-membered fused rings were studied. Considering of the carbon chain joining the oxime role to the ortho-carbon of the benzene band, the phenonium ion that normally facilitates phenyl migration may be unable to assume its preferred structure (three-membered band orthogonal to the phenyl ring). The three-carbon bridging chain for northward = 6 is such a case, and rearrangement of the anti isomer is very wearisome. As the length of the bridging concatenation increases, its constraint is less severe, and the rate of rearrangement increases. The eight-membered oxime picrate hydrolyzes quickly, producing a ix-membered lactam in high yield.
R2C=Due north-NH2 + HNOtwo ![]() RCONHR + Due north2
RCONHR + Due north2
Beckmann blazon rearrangements may also be carried out by treating hydrazones with nitrous acrid, equally shown on the right. As a rule, this is a less desirable process considering pure hydrazones are more hard to acquire, and the yield of pure product is inferior.
A straight rearrangement of ketones, thereby avoiding the necessity of preparing an derivative, is possible past a procedure known equally the Schmidt rearrangement. Acid-catalyzed addition of hydrazoic acid to the carbonyl group of a ketone creates an unstable azidocarbinol that, on dehydration, produces the same triazonium cation presumably formed as an intermediate in the nitrous acid deamination of a hydrazone. Rearrangement of this species past rapid nitrogen loss then initiates a Beckmann-similar rearrangement. Past clicking the upper diagram a second time, 2 examples of the Schmidt rearrangement will be presented.
The Stieglitz Rearrangement
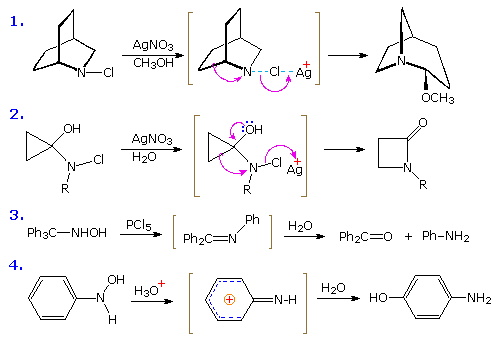
Examples of rearrangements to spii hybridized azacations are relatively rare compared with their carbon analogs. The starting materials for generating such azacations are usually chloramines or hydroxyl amines. Four examples of these transformations, sometimes called "Stieglitz rearrangements", are shown below. The first case is similar to a Wagner-Meerwein rearrangement, and the 2nd to a pinacol rearrangement. Competitive shifts of para-substituted phenyl groups in reactions similar to example #3, demonstrate that a methoxy substituent facilitates rearrangement, whereas a nitro substituent retards it. In the last example, a conjugated azacation activates the benzene ring to nucleophilic substitution, in contrast to the usual part played by amine substituents.
Rearrangement of Acyl Nitrenes to Isocyanates
Several useful and general procedures for degrading carboxylic acid derivatives to amines all involve the rearrangement of an acyl nitrene to an isocyanate. Although the nitrogen atom of a nitrene has no formal charge, it is electron deficient and serves as a locus for 1,2-rearrangements. Equally illustrated in the following diagram, acyl nitrenes may be generated from unlike amide-like starting compounds. One time formed, acyl nitrenes quickly rearrange to relatively stable isocyanate isomers, which may exist isolated or reacted with hydroxylic solvents. The about common application of this rearrangement is for the synthesis of amines. Thus, addition of water to the ketene-like isocyanates produces an unstable carbamic acid that decomposes to an amine and carbon dioxide. Full general procedures for obtaining the nitrene precursors are listed below the diagram.
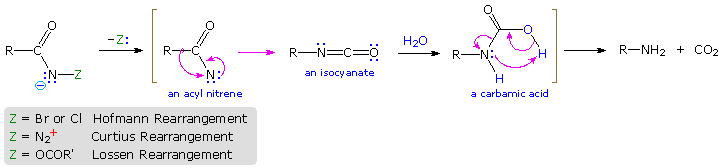
Hofmann Route: Primary amides are converted to N-halogenated derivatives past the activeness of HOX or Xii in alkali metal solution. Excess base generates a conjugate base of the product.
Lossen Route: A hydroxamic acid derivative (RCONHOH) is made by reacting an ester with hydroxyl amine. The hydroxamic acid is O-acylated and so converted to its conjugate base of operations.
Curtius Route: An acyl azide (RCON3) is prepared in one of two ways. (i) Reaction of an acyl chloride with sodium azide, or (ii) Reaction of an ester with excess hydrazine, followed by reaction of the acylhydrazide product (RCONHNHtwo) with cold nitrous acid. Acyl azides decompose to isocyanates on heating.
Schmidt Road: A variant of the Curtius procedure in which a carboxylic acid is heated with hydrazoic acrid (HN3) and an acid goad.
Some examples of these unlike rearrangements are shown in the following diagram. In each instance a light-green capitol letter (C, H, L or Due south) designates the blazon of reaction. The showtime 3 reactions illustrate the Hofmann rearrangement, which is a particularly useful method for preparing amines. The last case shows a Curtius reaction practical to a diester by style of an intermediate bis-acylhydrazide. An alternative Curtius approach to the amine product of case # 2 is besides shown. The Curtius procedure is particularly useful if the isocyanate is the desired product, since the decomposition of the acyl azide intermediate can exist conducted in the absence of hydroxylic solvents that would react with the isocyanate.
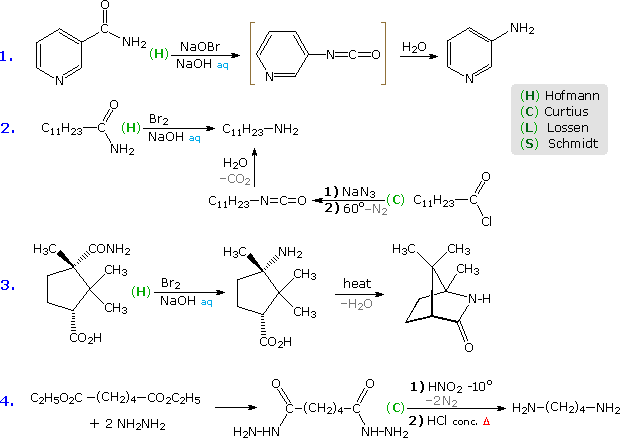
Five more examples will be displayed above by clicking on the diagram. Example # 5 shows a Schmidt reaction in which an optically active carboxylic acid is the substrate. The Due south-configuration of the migrating phenethyl grouping is retained in the amine product, confirming the intramolecular character of these rearrangements. Retention of configuration is also observed in Curtius, Lossen and Hofmann rearrangements of this kind. Reactions # six & # 7 are interesting cases in which water is absent during the formation and reaction of the isocyanate. The alcohol solvent in # 6 adds to the isocyanate to produce a carbamate ester, known equally a urethane. Unlike the unstable carbamic acids, urethanes do non decompose and may be isolated every bit pure compounds. If water had been the solvent, the resulting 1º-enamine would have rearranged to an imine and hydrolyzed to an aldehyde. In the Lossen rearrangement (# 7) butyl amine adds to the isocyanate to give a substituted urea (urea is NH2CONH2).
The Hofmann rearrangement in reaction # viii provides a novel example of the tautomerism of an acetylenic 1º-amine to a nitrile. Finally, the last instance illustrates a selective Hofmann rearrangement of a bromo-imide. The reactivity of the carbonyl group para to the electron withdrawing nitro substituent is increased relative to the other imide carbonyl. Consequently, base-catalyzed hydrolysis takes place in that location preferentially, leaving the acyl nitrene moiety meta to the nitro office.
Rearrangements of Acyl Carbenes |
|---|
Rearrangements of Acyl Carbenes
i. The Arndt-Eistert Reaction
The rearrangement of acyl nitrenes to isocyanates that is the crux of the Hofmann, Curtius and Lossen rearrangements, is paralleled by the rearrangement of acyl carbenes to ketenes, a transformation called the Wolff rearrangement. This rearrangement is a disquisitional step in the Arndt-Eistert procedure for elongating a carboxylic acid by a single methylene unit, as described in the diagram beneath. The starting acrid, written on the left, is converted commencement to an acyl chloride derivative, and and then to a diazomethyl ketone. Diazomethane has a nucleophilic methylene grouping, equally indicated past the resonance formulas drawn in the shaded box. Acylation of the methylene carbon produces an equilibrium mixture of a diazonium species and the diazomethyl ketone plus hydrogen chloride (written in brackets). If the HCl is not neutralized by a base of operations, this mixture reacts further to give a chloromethyl ketone with loss of nitrogen. However, if the HCl is neutralized as it is formed, the relatively stable diazo ketone is obtained and may be used in subsequent reactions.
Since diazomethane itself may part equally a base of operations, the grade of a given reaction is established by the manner in which the reactants are combined. When an ether solution of diazomethane is slowly added to a warm solution of the acid chloride, nitrogen evolution is observed and the chloromethyl ketone is the principal product. 1 equivalent of diazomethane is required for this reaction. If the addition is reversed, so that a cold solution of the acrid chloride is added slowly to an backlog of diazomethane in common cold ether solution, nitrogen development is once more observed; merely two equivalents of diazomethane are consumed. The products are the diazo ketone and methyl chloride (a gas) from the reaction of diazomethane with HCl..
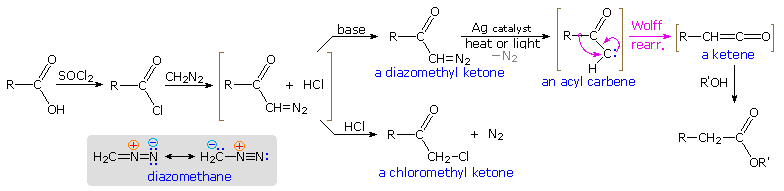
To deport out the Arndt-Eistert reaction the diazo ketone is decomposed in the presence of a silver catalyst (usually Agone2 or AgNO3 ) together with heat or light energy. The resulting Wolff rearrangement generates a ketene, which quickly reacts with whatsoever hydroxylic or amine reactants that may be present in solution. The full general equations on the left below illustrate that the cease product from the Arndt-Eistert reaction may be a carboxylic acid, an ester or an amide.
| 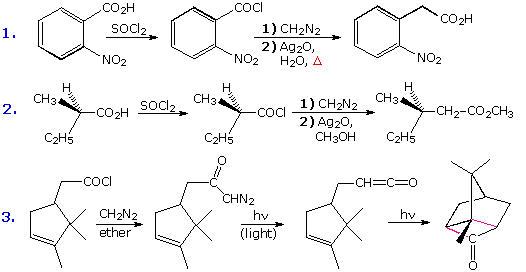 |
Three specific examples of this process are presented to the right of the general equations. The starting time ii examples are typical Arndt-Eistert reactions. Tolerance for other functional groups, such equally nitro, is demonstrated by the first case; and the second instance shows that the configuration of the migrating grouping is retained in the rearrangement. Other ketene reactions, such equally a [2 + 2] cycloaddition, may have place in the absenteeism of hydroxylic solvents or amines. Reaction #iii is an case of such an alternative reaction. The new bonds in the cycloadduct are colored pinkish.
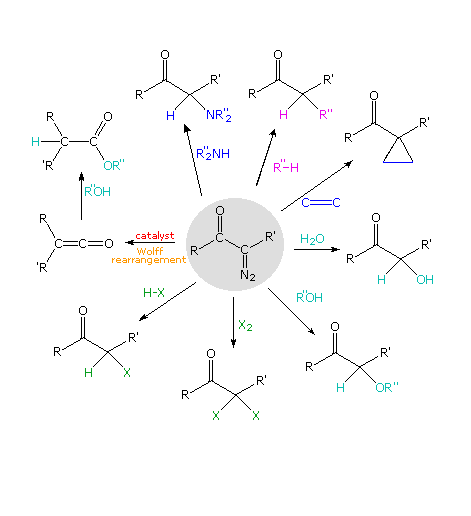
2. Diazo Ketone Reactions
The Arndt-Eistert reaction is a special instance of a more full general class of diazo ketone reactions. If nosotros presume that diazo ketones normally decompose to acyl carbenes, then numerous subsequent reactions can be imagined, and many accept been realized. The following diagram outlines some of these transformations, originating from the diazo ketone formula in the center of the diagram. Molecular nitrogen is lost in each case, and the Wolff rearrangement path is on the left. Most of the other reactions reflect the ability of carbenes to insert into sigma bonds or add to double bonds. Metallic catalysts are sometimes used to facilitate sure reactions, and may associate with carbenes to generate carbenoid intermediates.
By clicking on the diagram some examples of these diazoketone reactions volition be shown.
Exercise Problems |
|---|
| Some issues involving molecular rearrangements are provided here. |
This page is the belongings of William Reusch. Comments, questions and errors should be sent to whreusch@msu.edu.
These pages are provided to the IOCD to assist in chapters building in chemical teaching. 05/05/2013
millerovereatitese.blogspot.com
Source: https://www2.chemistry.msu.edu/faculty/reusch/virttxtjml/rearrang.htm
{kind=link}
Post a Comment for "From What You Know From Lecture Another Organic Product Is Almost Certainly Formed"